Research in the Rohani lab focuses on population biology, usually of host-natural enemy interactions, with a view to understanding fundamental processes in ecology and evolution. We use a combination of mathematical modelling, data analysis and statistical inference to understand the ecology and evolution of infectious diseases of humans and wildlife, including childhood infections and emerging infectious diseases.
Human and Wildlife Infectious Diseases
Much of current research in the lab is based on understanding long-term data sets on the spatio-temporal patterns of morbidity and mortality caused by microparasitic infections of childhood (such as measles, whooping cough and polio) and in wildlife systems (avian influenza viruses). The analyses of these data have provided interesting insights into the mechanisms of transmission and the ecology of infectious diseases.
Ecology, Immunology and Evolution of Pertussis
Historically, the study of pertussis has been a major focus in the lab and typifies the overall range of approaches we adopt to the study of infectious disease systems:
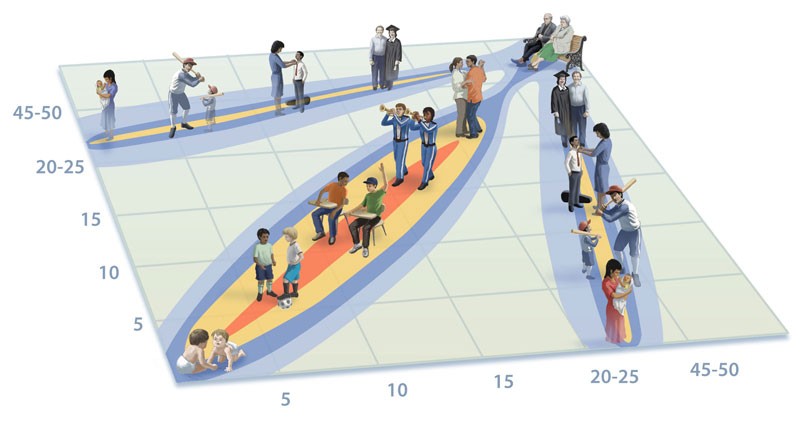
Illustration of age-specific patterns of contact, following Rohani et al. (2010; Science). Data based on Mossong et al. (2009; PLoS Med).
Spatio-temporal dynamics
We have been documenting the diversity of patterns in pertussis incidence using data from a number of sources. We have demonstrated how clear waves of pertussis incidence in the US during the 1950s gave way to spatially unorganized and irregular epidemics in the 2000s (Choisy & Rohani 2012). In contrast, we find no evidence for geographical pulsing in pre-vaccination era pertussis epidemics in England & Wales (Conlan et al., in prep), contemporary reports from the provinces of Thailand (Blackwood et al. 2013) or regions of Italy (Magpantay et al., 2016).
More recently, we have examined spatial synchrony in pertussis epidemics in the boroughs of London, identifying multi-focal recurrent waves (Saeidpour et al. 2022). To understand the potential drivers of these waves, we have used feed forward neural networks to identify potential borough-specific predictors of epidemic phase.
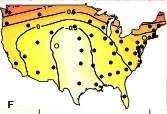
Waves of pertussis epidemics in the US from 1951-1962 (Choisy & Rohani 2012)
Nature and duration of immunity
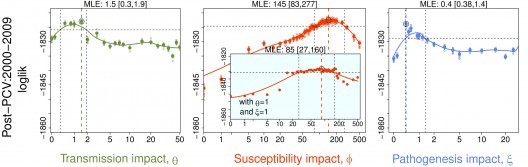
A key issue in the epidemiology of pertussis remains the duration of immunity, both derived following vaccination and natural infection. Using probe-matching approaches, Wearing & Rohani (2009), explored when model dynamics are in parsimony withepidemiological data. The main conclusion of this study was that mean duration of natural immunity may be in excess of 30 years. Last year, in the paper by Blackwood et al. (2013), we competed a range of models to explain contemporary pertussis incidence data from Thailand; we found long-term duration of immunity provided the most parsimonious explanation for the data. Recently, we have developed theory on the epidemiological consequences of different mechanisms behind immune “failure”, specifically the distinction between leaky and all-or-nothing immunity (Magpantay et al. 2014). Currently, we are in the process of evaluating the empirical evidence in support for these alternative explanations using computational statistics approaches on incidence data from England & Wales (Domenech de Celles et al., 2018) and Italy (Magpantay et al., 2015). In collaboration with Matthieu Domenech de Celles, we continue to examine this issue by studying data from Sweden, Denmark and the USA.
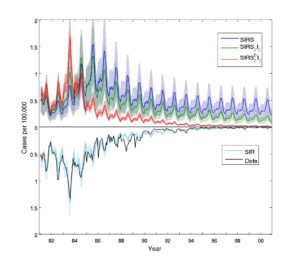
Comparison of different models with pertussis incidence data from Thailand (Blackwood et al. 2013; PNAS).
Designing effective booster vaccination schedules
Despite much uncertainty over the mechanisms that have shaped the resurgence of pertussis in countries such as the US and the UK, public health policy makers have had to implement responsive control strategies, in the shape of boosters introduced into the existing schedule. Given the complexity and high dimensionality of the problem, it isn’t well-suited to traditional optimization methods. So, we have been using a genetic algorithm approach to identify effective booster strategies under different assumed underlying causes of resurgence (Riolo & Rohani 2015).
Schematic of genetic algorithm used in Riolo & Rohani (2015; PNAS).
Pertussis immunization and diagnostics in a low-middle income setting
In collaboration with colleagues at Boston University, we have examined the adherence to routine immunization schedules (Gunning et al. 2020) and have established the usefulness of the full range of qPCR CT values in informing pertussis epidemiology (Gill et al. 2021). Additionally, this study has identified a high incidence of asymptomatic pertussis, notably among infants.
Multi-pathogen systems
Interaction between influenza and pneumococcal bacteria
Numerous studies focused at the level of the individual—including lab challenge studies on mice and autopsy reports on humans—have established that infection with the influenza virus affects the outcome of exposure to bacteria. In contrast, efforts to uncover a clear signal of this phenomenon in population-level incidence reports have been unequivocal. We have developed a likelihood-based hypothesis-testing approach using a mechanistic pneumococcal transmission model and identified a clear, unambiguous interaction, resulting from a short-lived but notable increase in susceptibility to bacterial pneumonia following influenza infection (Shrestha et al. 2013). Ongoing work will extend this to all-cause pneumonia (Shrestha et al. 2015).
Likelihood profiles testing alternative hypotheses regarding the impact of influenza infection on pneumococcal pneumonia. From Shrestha et al. (2013; Sci. Trans. Med.).
Mixed transmission systems
Over the past ten or so years, we have been working with David Stallknecht and John Drake (both at the University of Georgia) and Benjamin Roche (Montpellier) to understand the transmission dynamics of Influenza A viruses among their avian hosts. In particular, this work has focused on the consequences of environmental transmission via long-lived viral reservoir.
Transmission ecology
We have shown that indirect transmission chains can have important consequences for the invasion (Rohani et al. 2009), persistence (Breban et al. 2009) and coexistence of these viruses (Breban et al. 2010; Roche & Rohani 2010). In Delaware Bay, described as a global hotspot for avian influenza viruses, environmental transmission is shown to be a crucial component in the triple prevalence peaks reported during the year (Brown & Rohani 2012, Brown et al. 2013).
Virulence evolution
In collaboration with Benjamin Roche and John Drake, we have studied the impact of mixed transmission dynamics on the evolution of virulence, documenting the necessary conditions for evolutionary bistability (Roche et al. 2011).
Influenza phylodynamics
The study by Roche et al. (2014) documented contrasting patterns of viral genetic diversity among human and avian influenza viruses. Within comparable subtypes, we observe an order of magnitude greater HA diversity among avian viruses. We examined putative explanations and found that HA genetic diversity in avian viruses is determined by a combination of factors, predominantly subtype-specific differences in host immune selective pressure and the ecology of transmission (in particular, the durability of subtypes in aquatic environments). Our conclusion that environmental transmission plays an important role in the evolutionary biology of avian influenza viruses—a manifestation of the ecological phenomenon called the ‘‘storage effect’’—highlights the potentially unpredictable impact of wildlife reservoirs.
Tipping points in emerging and re-emerging infectious disease systems
In collaboration with John Drake (UGA), Andrew Park (UGA), Matt Ferrari (Penn St) and Bogdan Epureanu (Michigan), we received an NIH MIDAS grant to forecast tipping points in infectious disease systems — we have called this project AERO (Anticipating Emerging/Re-emerging Outbreaks). Our study developed new theoretical models to study the transmission of infectious diseases near tipping points (Brett et al. 2017, Brett et al. 2020, Drake et al. 2019).
Project on forecasting tipping points in emerging and re-emerging infectious diseases
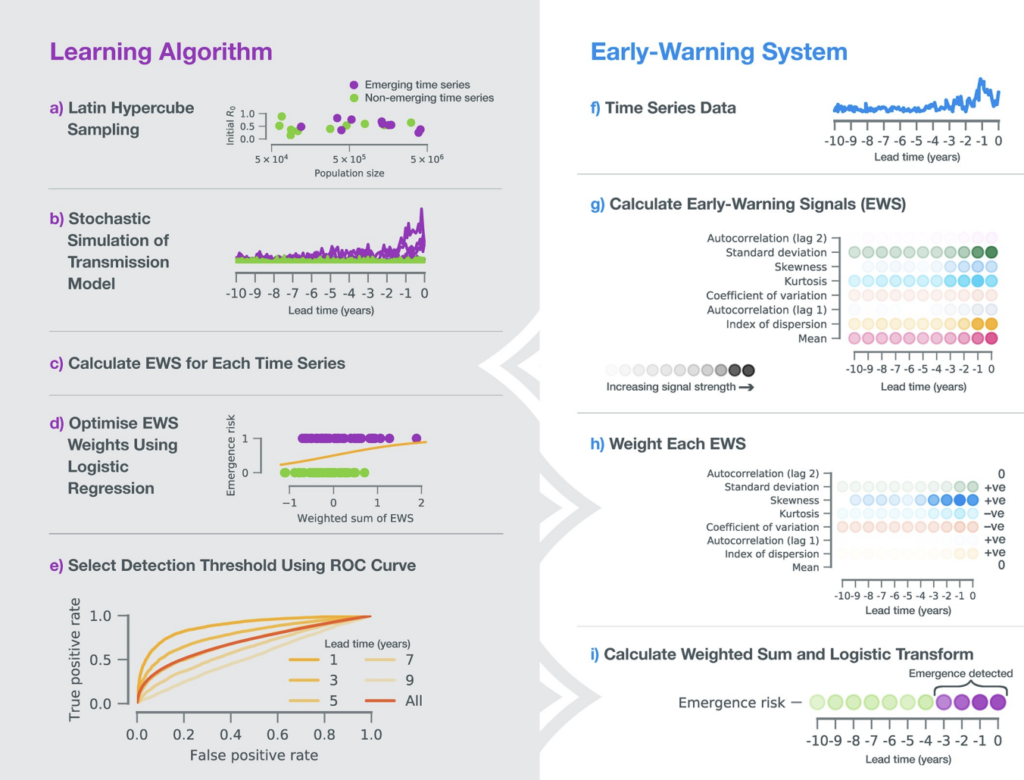
Having identified candidate statistical signatures of critical slowing down in both low and high demonsional models, we have been working on transfer learning algorithms for the detection of tipping points in epidemiological data. Our approach to emergence detection was published in Brett & Rohani (2020).
The figure on the right illustrates our pipeline for model training (left column) and early warning system (right column).
In April 2021, we were award an NIH Center of Excellence in Influenza Research and Response. Our Center for Influenza Disease & Emergence Research (CIDER) is comprised of colleagues from UGA (College of Veterinary Medicine, Odum School of Ecology, School of Public Health, Institute of Bioinformatics) as well as outside collaborators (St Jude Children’s Hospital, Rochester University, Boston Children’s Hospital and the University of Melbourne). CIDER will conduct research in three continents. The Rohani lab will contribute to the informatics mission of CIDER, starting with a research project in collaboration with Mark Tompkins on the within-host dynamics of influenza B viruses in ferrets and mice.
As part of CIDER, we have received an option to develop computational methods for influenza forecasting. In collaboration with John Drake, we are working on developing ensemble methods. In a separate project, we are exploring ways in which sequence-based algorithms can be incorporated into seasonal forecasting models.
Predicting Seasonal Influenza
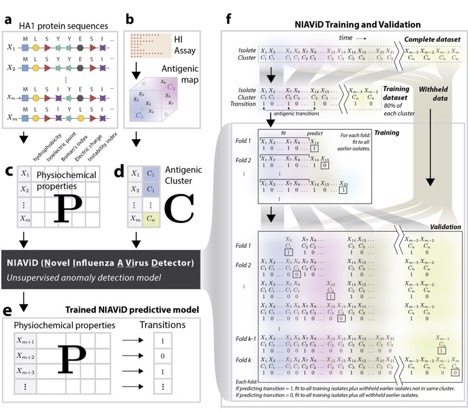
In collaboration with Justin Bahl, John Drake and others, we have been working on a CDC-funded project to predict influenza antigenic evolution. This work involves a combination of statistical learning approaches applied to HA sequences, viral physicochemical traits and indicators of immune response, including HI titres. In particular, lab postdoc Alpha Forna has developed an anomaly detection algorithm that uses traits estimated from sequences (eg, hydrophobicity, electrostatic charge etc) to classify sequences as novel or belonging to a prior antigenic cluster.
Figure on right depicts our pipeline for model development and testing platform.